
2023 专家共识:微移植治疗老年急性髓性白血病
白血病相关性口腔病损临床诊治的研究进展
血小板减少性紫癜的激素依赖问题

早预防早治疗,带你快速了解白血病!
关于血小板减少症:帮你答疑解惑!

MYH9相关血小板减少症
多发性骨髓瘤做了自体移植还会复发吗?
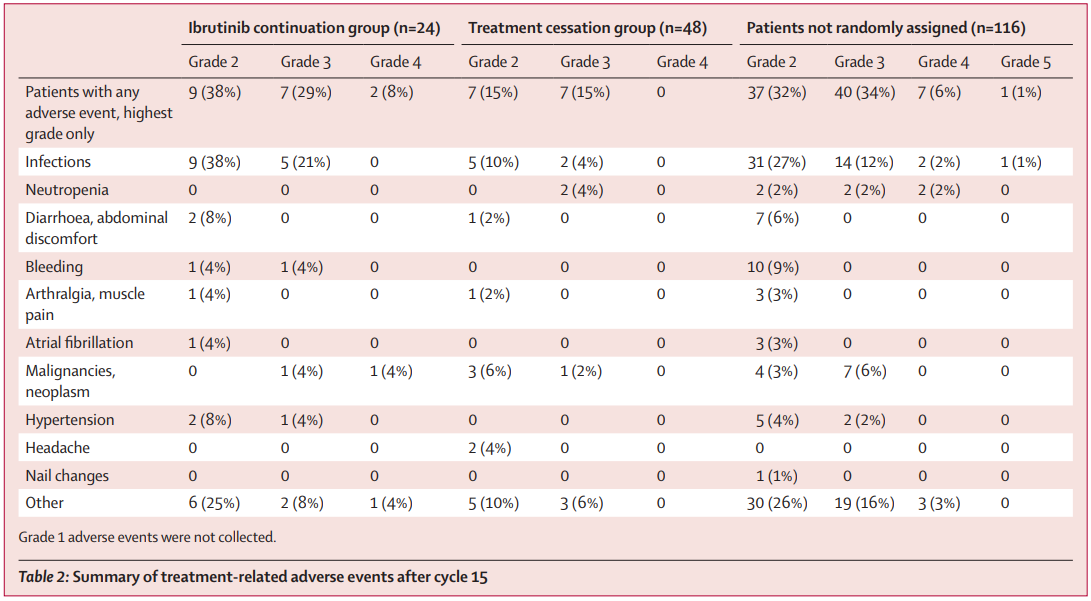
MRD或可指导复发/难治慢性淋巴细胞白血病患者治疗
缺铁性贫血的诊断标准
边缘区淋巴瘤的新药治疗
慢性淋巴细胞白血病的研究进展
艾曲泊帕-助力ITP患者安心生活
上一页
1
2
...
6
下一页

服务号

订阅号


无极血康中医医院 冀ICP备05003291号-1 网站建设:中企动力 石家庄 SEO